
About Enchondroma and Maffucci
General Information
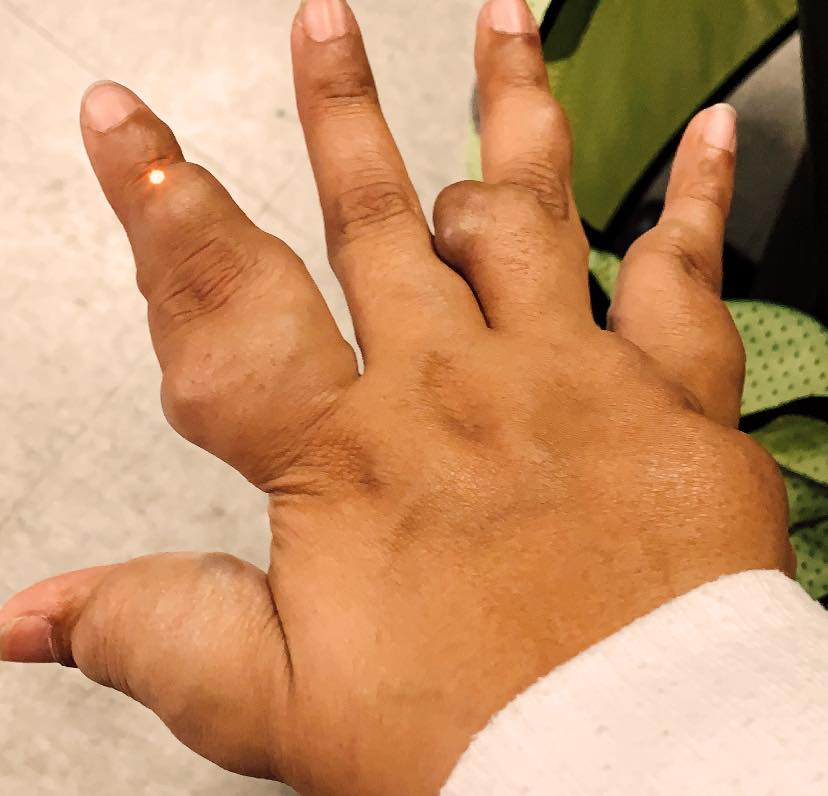
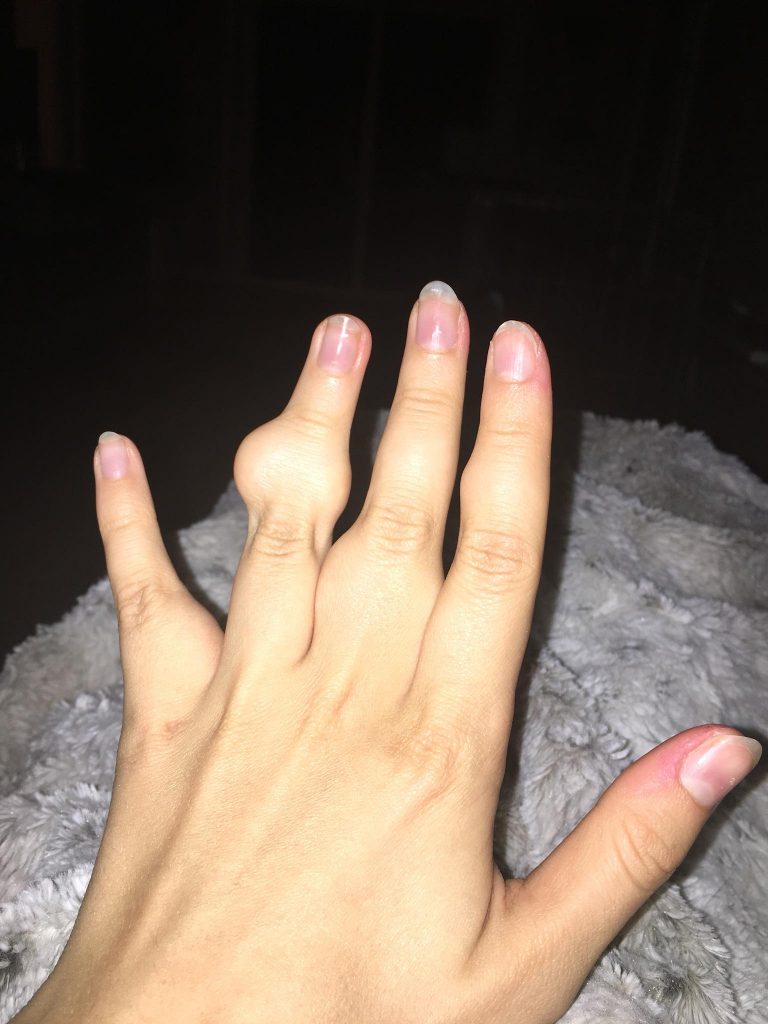
Ollier disease and Maffucci syndrome are cancer predisposition syndromes caused by germline or early postzygotic variants and subsequent hits in the same or different genes lead to formation of enchondromas, vascular anomalies, and chondrosarcoma. Most Ollier RAREsies are affected in the fingers, toes, and femur. Further awareness and research will help provide more answers. Ollier disease and Maffucci syndrome occur as isolated cases.
For more information please visit OMIM, using the following codes for Ollier disease: 166000 Maffucci syndrome: 614569
Ollier and Maffucci natural history is available at https://jhmi.co1.qualtrics.com/ife/form/SV 9v3S8w6UH1f5ur
Conclusions based on the review of the literature. The median age of the enchondroma diagnosis in RAREsies with Ollier disease was 4 years younger than RAREsies with Maffucci syndrome. Interestingly, Maffucci RAREsies were diagnosed with enchondromas at a median age of 11 years old, but they were often misdiagnosed as having Ollier disease since the median age of the vascular anomalies diagnosis was 13 years old.
The prevalence of chondrosarcoma among published RAREsies with Maffucci syndrome was ~31% and ~30% among RAREsies with Ollier disease. This prevalence is lower than the 45.8% for RAREsies with Ollier disease and 57.1% for RAREsies with Maffucci syndrome as previously described/noted. In these RAREsies, vascular anomalies were identified in internal organs only. The prevalence of malignant tumours among RAREsies with Maffucci was 48.7% and 53.1% among RAREsies with Ollier disease. Brain tumours were significantly more common among RAREsies with Ollier disease. Among RAREsies with Maffucci, the malignant tumours appear on average 15 years after the appearance of an enchondroma, at a median age of 30. Among RAREsies with Ollier disease, the malignant tumours appear in average 11 years after the appearance of an enchondroma, at a median age of 25.
Conclusions based of the survey’s responses. Among the RAREsies with Ollier we found that 53.1% had bilateral enchondroma, against 42.2% of the RAREsies analyzed in the literature review. We also found that 23.8% of the RAREsies with Ollier disease had the axial skeleton affected similarly to the RAREsies in the literature review. Ribs, spine, scapula were found to be affected. The prevalence of chondrosarcoma among RAREsies with Maffucci syndrome was ~30% and ~22% among RAREsies with Ollier disease, similar to what we found with the literature review and also lower than the ones previously described. The prevalence of malignant tumours among RAREsies with Maffucci syndrome was 36.1% and 25.4% among RAREsies with Ollier disease. The median age of disease onset was 22 years among RAREsies with Maffucci syndrome and 28.5 years among RAREsies with Ollier disease. In our findings, malignant tumours are less common among RAREsies with Maffucci than the RAREsies described in the literature (36.1% against 48.7%), and RAREsies were diagnosed with Maffucci in a younger age (22 against 30). The RAREsies with Ollier disease had a lower prevalence of malignant tumours than the RAREsies with Maffucci syndrome. The median age of Maffucci syndrome RAREsies that took part in the survey is 39.7, the median age of Ollier disease RAREsies is 17.6. The survey also guided us to unexpected diagnoses in these RAREsies such as D-2-hydroxyglutaric aciduria 2, metachondromatosis, and multiple exostoses.
Diagnosis and Surveillance
Protocol for Ollier and Maffucci
Patients should be submitted to a full skeletal survey at the time of their diagnosis for a full assessment of all bones for the kind of bone/or cartilage tumours they may have. Depending on the type of bone and cartilage lesions that they have, on the other features that they present, and on their family history, sequencing of PTPN11, EXT1, EXT2, EXT3, ACP5 (enchondroma), and/or COL2A1 (Maffucci) is indicated. Deletions and duplications should of course be investigated by an array. A urine organic acid test should be performed for investigation of hydroxy glutaric aciduria. In children, periodic clinical screening every 6-12 months should be performed for early treatment of growth abnormalities.
Ollier & Maffucci Natural History.
We collected data from 287 publications describing individuals with Ollier disease and 199 individuals with Maffucci syndrome.
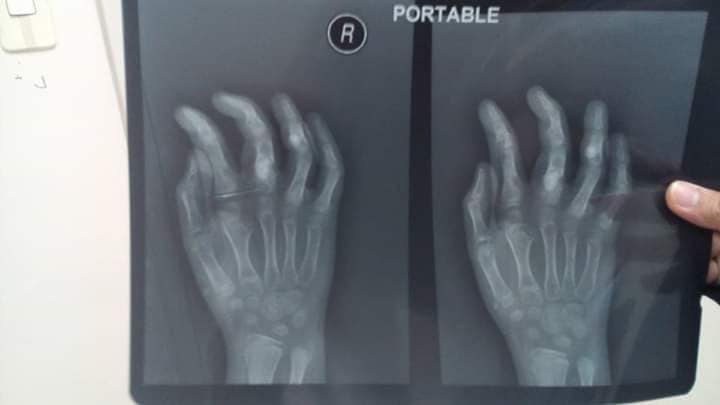
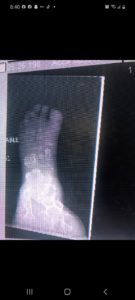
Radiographic Presentation
- Localized, radiolucent defect usually punctuate calcifications
- calcifications are typical, but not always present
- Matrix may deomonstrate various degrees of calcification
- Calcifications are stippled, punctate, popcorn like calcifications and “Ring and Arc” calcifications
- Cartilage tumors grow in a lobular manner. The perimeters of the lobules undergo enchondral ossification that may calcify. If the entire perimeter of the lobule calcifies it appears radiographically as a “Ring”. If a portion of the perimeter of a lobule calcifies it forms an “Arc” on an x-ray
- May be located centrally or eccentrically
- Grows eccentrically or concentrically (phalanges)
- Cortex may be scalloped and thinned in the phalanges
- MRI is better to see noncalcified chondroid lesions, and the full extent of lesions
- Most commonly found in metaphysis
Plain X-Ray
- Geographic lytic lesion
- Central, often metaphyseal in long bones. May also be eccentric
- Expansile remodeling with thinned cortex
- Chondroid matrix with calcifications in majority of tumors
- Approximately 20% ahve limited, or no calcifications
- MRI
- Lobulated margin
- Marked increased intensity long TR images
- Calcified chondroid – low intensity all sequences
MRI:
- Lobulated margin (Lobular Growth Pattern)
- T1 Weighted Images: Intermediate Signal Intensity
- Calcifications will be low signal
- T2 Weighted Images: High Signal Intensity
- High water content shows as high signal on T2 weighted images
- Calcifications will be low signal
- Marked increased intensity long TR images
- Calcified chondroid – low intensity all sequences
- There should never be any cortical destruction nor a soft tissue component. If this exists then the tumor must be a chondrosarcoma.
- Endosteal scalloping and cortical expansion is acceptable for phalangeal tumors. In most benign long bone cartilage tumors there is minimal endosteal scalloping but there should be no cortical expansion nor thickening. There should be no cortical destruction and no soft tissue component associated with an enchondroma. Cortical destruction, periosteal thickening, cortical expansion and a soft tissue component indicates a chondrosarcoma of the long bone.
Pathology
- Rests of hyaline cartilage with a lobular growth pattern
- Cells are within lacunae
- Hypocellular with cells spaced apart separated by matrix
- Cells have small nuclei and are similar size and shape (no pleomorphism)
- No mitotic figures
- The matrix has a ground glass basophilic appearance
- The matrix contains glycosaminoglycans that attract fluid/water and gives it a bsophilic appearance and show as high signal intensity on T2 weighted MRI
- The collagen of the matrix is organized in a manner such that the refractile index under a microscope gives a ground glass appearance to the matrix. One can not visualize the actual collagen fibers.
- Hyaline cartilage often with myxoid areas
- Variable amorphous calcification and enchondral ossification
- May cause mild expansile remodeling of bone and cortical thinning
Gross Pathology
- Fragments of enchondroma after curettage are bluish white and glistening hyaline cartilage
- There may be yellow calcified foci
Cartilage grows in a lobular manner and hence in a completely resected specimen there will be local lobules of mature cartilage
Microscopic Pathology
- Enchondromas are well defined lesions with cartilage arranged in lobules that are separated by fibrovascular septa
- Enchondral ossification may occur around periphery of lobules and when calcified appear as “Rings and Arcs” on X-rays
Cells are in lacunae and have small dark nuclei
Low cell count, cells appear bland with few chondrocytes and are similar size and shape
- Although some enchondromas may have areas that are hypercellular and may have two or three cells within a lacunae
No entrapment or destruction of trabeculae
There should be no myxoid change in long bone lesions (there is occasional myxoid change in enchondromas of the digits)
Presence of myxoid change is sign of malignancy
Enchondromas of digits can have hypercellularity, bi and trinucleated cells and myxoid change and still be considered benign
Different Diagnosis
- The main differential is with a low grade (Grade 1) chondrosarcoma
- Can be very difficult to differentiate from a Grade 1 Chondrosarcoma from an enchondroma based on histology
- It is easy to differentiate from a grade 2 or 3 chondrosarcoma by means of histology which are hypercellular, pleomorphic and demonstrate mitotic figures
- Radiographs and clinical history are important for differentiation
Biopsies are not useful for differentiating an enchondroma from a low grade chondrosarcoma
- Pain attributable to lesion
- Age greater than 50
- Cortical destruction and a soft tissue mass
- Periosteal reaction and thickening
- Endosteal erosion>2/3 cortical thickness on a CT scan
- Size greater than 5 cm
Bone Scan: Lesion that is hotter than ASIS
Treatment
- Enchondromas are benign, indolent (not growing) tumors
- Indications for surgery:
- Digits: Impending or actual pathological fracture
- Intralesional curettage and bone graft or cement
- Long bones: Rare to fracture usually observe
If growing it is considered chondrosarcoma and would recommend surgery accordingly
- Digits: Impending or actual pathological fracture
Prognosis
- Recurrence rate following curettage is <5%
- Recurrence of an enchondroma suggests malignancy
- There are rare cases where enchondromas can dedifferentiate into a chondrosarcoma or dedifferentiated chondrosarcoma (low grade chondrosarcoma with an osteosarcoma, fibrosarcoma or malignant fibrous histiocytoma arising adjacent to it)
Enchondromatosis
- (Multiple Enchondromas/Ollier Disease
Enchondromatosis is a rare disorder that is not hereditary in which the patient is afflicted with multiple intraosseous cartilaginous tumors or enchondromas.
Clinical Data
- Mild male predilection
- Present in childhood
- Usually affects the extremities
- Variable severity
- May be predominantly unilateral or affect a single extremity/limb
- Affected limb is often shortened and deformed and angulated
- May become stable at puberty
- Higher risk of malignant transformation to chondrosarcoma (5-50%) as opposed to an isolated enchondroma
- Marked skeletal deformity
- Not hereditary